
研究背景
太阳能驱动水分解,可以将太阳能转化为清洁可再生的氢燃料,而被认为是解决环境污染和能源问题最有效的途径之一。在众多半导体材料中,CdS的能带结构对于光催化分解水制氢的半反应是非常适合的。然而,由于其易光腐蚀、光生电子-空穴易复合等问题,阻碍了其进一步应用。相比之下,TiO2结构和性能非常稳定,可以对CdS进行保护,减缓其被光腐蚀。两者复合而成的异质结构,可显著地提高其使用寿命和催化效果。
成果简介
近日,best365官网登录入口化学化工学院刘志勇课题组在Chemical Engineering Journal上报道了一种SiO2牺牲模版法制备空心核壳结构的异质结光催化剂并用于光催化析氢,题为Synthesis of hollow core-shell CdS@TiO2/Ni2P photocatalystfor enhancing hydrogen evolution and degradation of MB(DOI:doi.org/10.1016/j.cej.2018.11.211),同时该文章应邀参选该期的封面。在这项工作中,他们结合了异质结、催化还原表面分离的优点,得到了一种CdS@TiO2/Ni2P复合光催化剂。首先以SiO2为模版,用CdS进行包覆。继而,将锐钛矿型TiO2包覆在外层作为壳层,并通过NaOH将SiO2刻蚀掉,从而获得了空心核壳结构的CdS@TiO2。CdS作为核层可以对可见光吸收,空心核壳结构可以增加光吸收作用,同时TiO2可以减少CdS的光腐蚀。最后,通过高温煅烧磷化的方法负载了5%wt的Ni2P作为助催化剂。通过对复合光催化剂产氢的性能和机理的研究,作者发现光生电子向壳层传输,并集中在外表面作为产氢位点的Ni2P上,从而还原水产生氢气,而空穴传输向核层与CdS的内表面,与牺牲剂反应。该研究提出氧化和还原过程空间分离的新型设计策略,为提升光解水产氢效率提供了新思路。刘志勇教授为本文通讯作者,博士研究生吴可量作为第一作者。
图文导读
| |
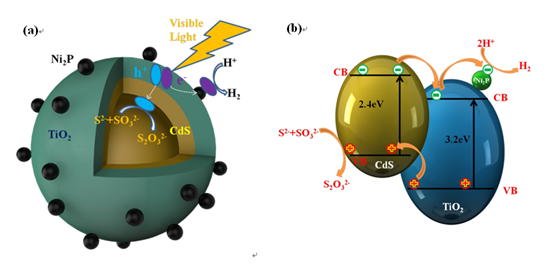
图1.(a)太阳光下CdS@TiO2/Ni2P核壳光催化体系电荷转移示意图。(b) CdS和TiO2的能级信息
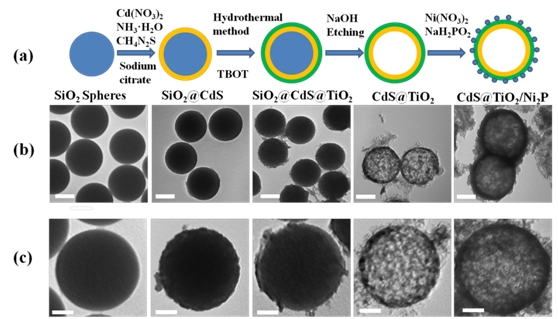
图2(a) CdS@TiO2/Ni2P制备原理图。(b)和(c) SiO2、SiO2@CdS、SiO2@CdS/TiO2、CdS/TiO2和CdS@TiO2/Ni2P的TEM图像(从左至右)。(c)和(b)中的比例尺分别为200和100 nm。
要点解析
图1为纳米复合催化剂的催化机理图,以及所用半导体材料的价带导带示意图。众所周知,纳米材料的微观结构决定了其性能,在CdS核层上包覆TiO2壳层,并使用非贵金属Ni2P作为助催化剂,有利于光生电子和空穴的分离。在太阳光照射下,电子和空穴由CdS核层和TiO2壳层共同激发产生。然后,电子转移到壳层外的Ni2P上(Ni2P的还原电位更低)。同时,由于CdS的氧化电位较高,空穴从TiO2迁移到CdS核层的内表面,并结合牺牲剂。而迁移到外表面的电子则参与了析氢反应。从反应动力学的角度来看,空心CdS@TiO2/Ni2P产生两个氧化还原空间分离的表面,是H2生成的理想光催化体系。

图3 (a) CdS@TiO2/Ni2P的低分辨率和高分辨率SEM图。(c) CdS@TiO2/Ni2P的EDX谱。(d) SEM图像及其元素分布图。比例尺为50nm。

图4. CdS@TiO2/Ni2P高分辨透射电镜
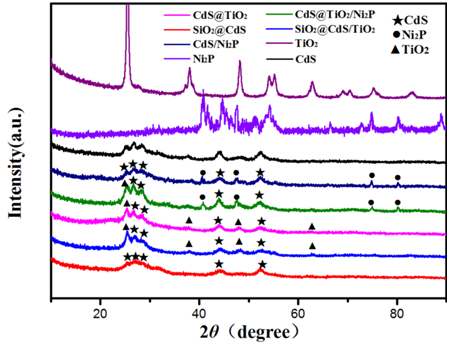
图5.样品的XRD图
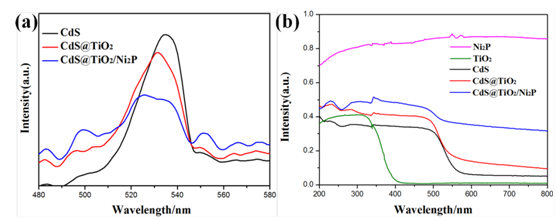
图6. (a) PL光谱图 (b)紫外吸收光谱图
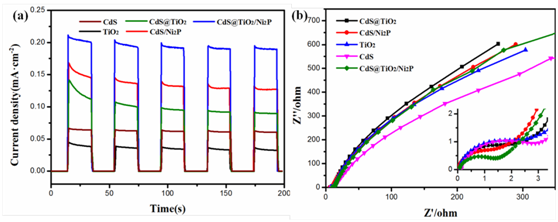
图7. (a)光电流图 (b)电化学阻抗图
要点解读
通过图6(a)可以看出激发出的电子复合程度为CdS@TiO2/Ni2P < CdS@TiO2 < CdS,说明复合催化剂有效地抑制了电子和空穴的复合。6(b)可以看出由于Ni2P带隙为1.0eV,所以在整个200-800nm的范围都有光吸收,TiO2的吸收带边为385nm的紫外区域,而CdS的吸收带边为530nm,形成复合催化剂后的吸收带边大约为720nm。对应在图7(a)中复合催化剂的光电流值也比复合前的值要大,7(b)中的阻抗要小一些,说明复合后的电阻是下降的。
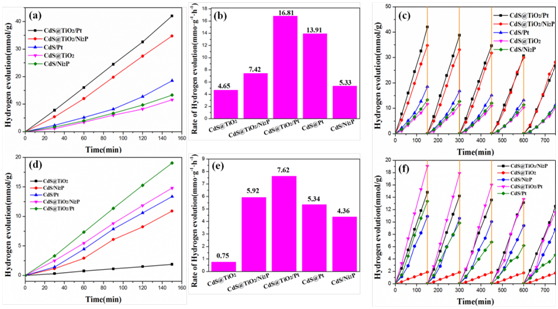
图8. (a)、(d)光催化制氢首次使用和(c)、(f)循环使用性能图。(b)、(e)催化剂第一次使用时的氢释放速率。(a)、(b)及(c)使用带有AM1.5G滤光片的氙灯光源,(d)、(e)及(f)使用带有420 nm截止滤光片的氙灯光源。
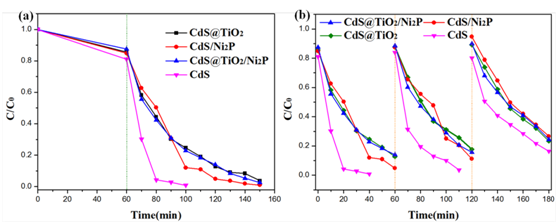
图9.以MB为模型底物,在模拟太阳光照射下,(a)不同催化剂的第一个循环内及(b)循环稳定性。
要点解读
不同样品的光催化析氢性能如图8所示。与CdS@TiO2/Ni2P相比,CdS@TiO2的2.5h析氢速率明显更低。TiO2存在于CdS核心外部,产生致密的界面,为电子/空穴复合提供了有利的位置,这通常会降低光析氢的效率。相比于CdS/Ni2P, CdS/Pt样品的产氢量略高,这可能是因为负载Pt有效地作为活性位点提取了电子,但相比于制备成本,Ni2P远低于Pt。在析氢过程中,作者还对所有样品进行了稳定性测试。CdS@TiO2/Ni2P和CdS/Ni2P的活性在5个周期后没有变化。然而,CdS@TiO2/Pt和CdS/Pt表现出催化活性递减后复苏周期,这很可能是由于Pt是光沉积在催化剂的表面,而Ni2P是在Ar流保护270°C下煅烧4 h负载上 (参见图8 C、f)。从HRTEM图像中可以看出,煅烧后Ni2P和TiO2晶格相交形成异质结,结合力更强。Pt仅还原沉积在表面,结合力较弱。由于空穴电子的快速重组和活性位点的缺乏,未负载助催化剂的CdS@TiO2作用下,氢气的析出量非常低。由于光腐蚀,所有样品在4次循环后,其光催化活性实际上略有降低。CdS@TiO2/Ni2P和CdS@TiO2/Pt样品经过5次循环后,氢气产率分别下降~18%和~38%,说明CdS@TiO2/Ni2P光催化剂的稳定性较好。
与此同时,作者还利用氧化还原空间分离光催化剂来降解亚甲基蓝(MB)(见图9a)。与CdS@TiO2相比,CdS@TiO2/Ni2P的光催化氧化活性略有增强,说明空间分离的还原面不利于氧化反应。与纯空心CdS相比,CdS@TiO2的活性较低,说明空穴和羟基自由基等氧化活性物质主要存在于CdS@TiO2外壳的内表面。然而,暴露在TiO2外壳外表面的空穴更容易与MB分子发生反应。因此,CdS@TiO2和CdS@TiO2/Ni2P的MB降解速率均低于纯空心CdS。但空间分离的CdS@TiO2/Ni2P仍具有光催化稳定性等优点。由于光腐蚀,CdS和CdS/Ni2P的光催化活性逐渐降低,且随着循环次数的增加,其活性逐渐降低(见图9b)。从单辅催化剂光催化体系来看,由于CdS表面剩余电荷的光腐蚀,MB的降解速率略有下降。正如预期的那样,CdS@TiO2/Ni2P由于具有TiO2的保护作用,即使经过三次循环也具有非常可靠的光氧化作用。
总结与展望
综上所述,本文作者采用硬模板法结合分步化学合成方法,成功合成了一种以CdS为核层,TiO2为壳层的新型氧化还原表面空间分离的CdS@TiO2/Ni2P光催化剂。与负载单一的助催化剂的CdS (Pt或Ni2P),空间上分离的设计显著提高光催化产氢能力 (模拟太阳光下为34.76 mmol g-1;在可见光下为14.80 mmol g-1)和循环稳定性(五次循环后仍具有:28.12 mmol g-1)。MB的降解率在第1个循环后为85%,3个循环后为77%。本文提出一种空心结构氧化还原面空间分离的策略,并结合非贵金属助催化剂,从而实现低成本、高效能光催化析氢的目的,为此类光催化剂的开发开辟了新途径。